Comprehensive Overview of Reticular Dysgenesis: History, Clinical Profile, Genetic Insights, and Treatment Advances
Posted on January 2, 2024 • 6 minutes • 1068 words
Table of contents
History
In 1959, De Vaal and Seynhaeve first documented a case of severe combined immunodeficiency (SCID) accompanied by congenital absence of white blood cells. This discovery was based on observations of monozygotic twin boys, born prematurely, who had no white blood cells at birth. Post-mortem examination of these infants, who unfortunately died from septic complications as neonates, showed conditions consistent with SCID. This included the absence of lymphoid cells in their lymphoid organs and myeloid cells in their bone marrow, while erythroid cells and megakaryocytes were present. This unique combination of symptoms led the researchers to name the condition “reticular dysgenesis” (RD), attributing it to a failure in the development of early multipotent reticular cells into myeloid and lymphoid cells . Further studies on RD revealed variations in white blood cell counts among patients, with some exhibiting immature myeloid cells up to the promyelocyte stage. Roper et al.’s extensive study, using cell-surface marker staining, indicated that RD might interfere with the late differentiation of lymphoid and myeloid cells. The first successful bone marrow transplantation (BMT) in 1983, which treated a patient with RD, pointed to the disease originating in stem cells rather than the bone marrow environment. RD patients were found to not respond to G-CSF treatment for granulopoiesis, unlike those with other congenital neutropenias. Additionally, primary sensorineural deafness emerged as another characteristic of RD, with adenylate kinase 2 deficiency identified as its molecular basis.
Clinical and Laboratory Presentation
RD patients are distinguished by several unique clinical features from other SCID forms:
- Low birth weight due to prematurity and/or intrauterine growth retardation.
- High fatality rate from neonatal infections.
- Severe blood leukopenia and agranulocytosis.
- Normal or decreased red cells.
- Normal or decreased thrombocytes.
- Normal or decreased marrow cellularity.
- Arrest in myeloid cell maturation in bone marrow.
Patients with RD typically seek medical help in the neonatal stage due to suspected infections or immaturity signs. A review of 16 RD cases highlighted a trend of low birth weights, often caused by both prematurity and intrauterine growth retardation. This contrasts with other SCID variants, where babies are usually born at term without growth retardation. Some RD patients also exhibited hepatomegaly or splenomegaly, and a few required surgical intervention for abdominal issues like intestinal stenosis. Post-transplantation, bilateral sensorineural deafness was commonly observed. Leukocytopenia and agranulocytosis are primary laboratory findings in RD, with automated WBC counting sometimes overestimating leukocyte counts due to the presence of nucleated red cell precursors. Anemia and thrombocytopenia were also noted in several cases, with no clear link to septic complications. The presence of maternal T cells in RD, often leading to graft-versus-host disease, was a common finding, particularly in contrast to other SCID variants.
Molecular Biology of RD
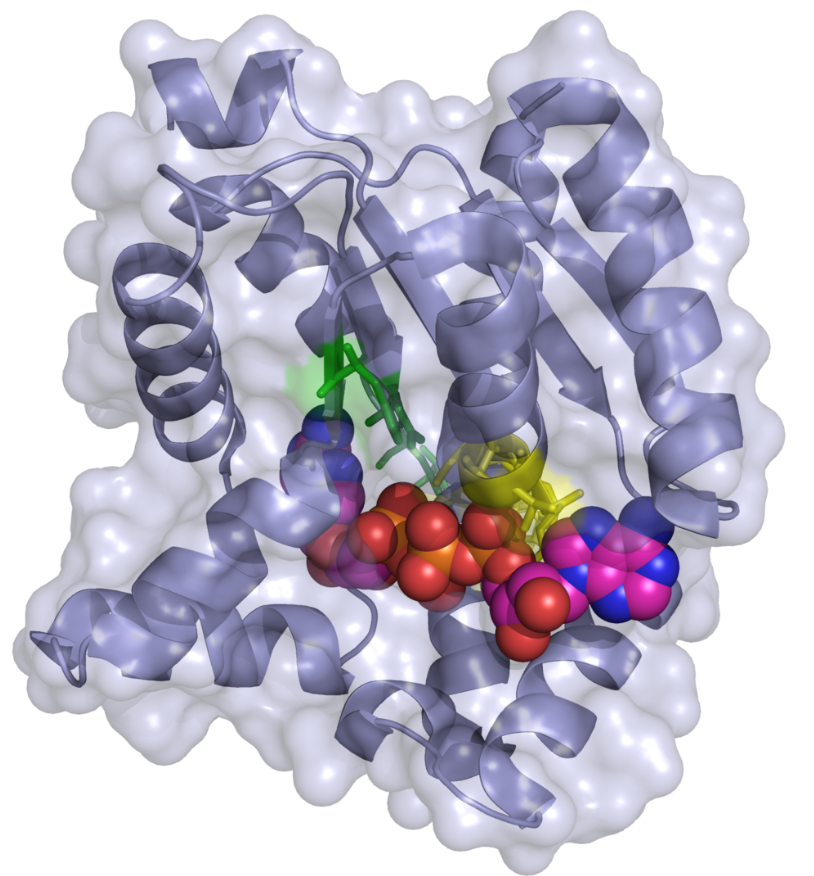
The gene responsible for RD, adenylate kinase 2 (AK2), was identified through homozygosity mapping and gene sequencing. The AK2 gene, which is highly conserved across species, is located on chromosome 1p35.1 and spans approximately 24 kb. It expresses two mRNAs, isoforms A and B, across various tissues (except erythrocytes), though their expression levels vary. Surprisingly, AK2 protein was found in the cochlear stria vascularis capillaries and blood vessels in mice, suggesting a potential role as an ecto-enzyme. The two isoforms differ slightly in their C-terminal amino acid composition. AK2, a mitochondrial protein, plays a crucial role in cellular energy metabolism and is involved in a novel apoptotic pathway, making it a subject of interest in molecular dynamic studies.
Mutation Analysis of RD Patients
Genetic analysis confirms RD as an autosomal recessive disorder caused by various mutations in the AK2 gene. These mutations include deletions, splice-site mutations, and missense and nonsense mutations, often inherited homozygously due to family consanguinity. Each family affected by RD typically has unique mutations, with no common ancestral mutation identified. Interestingly, these mutations do not seem to affect AK2 RNA levels. However, they significantly reduce protein stability, with most mutations resulting in a complete loss of AK2 function. A few cases of RD without identifiable AK2 mutations suggest the possibility of other genetic causes.
Animal Models of AK2 Deficiency
Research on zebrafish and drosophila has been pivotal in understanding AK2’s role. In zebrafish, a human AK2 mutation was simulated, leading to a nonfunctional protein. This mutation did not affect overall embryo development or erythropoiesis but led to the absence of developing lymphocytes, indicating Ak2’s specific role in leukocyte development. In drosophila, homozygous Ak2 mutants, developed normally until the larval stage, then ceased growth and died, suggesting that maternally provided AK2 supports early development but is insufficient later. These animal models, while differing from human phenotypes, provide valuable insights into AK2’s functions and its role in developmental processes.
Treatment
Hematopoietic stem cell transplantation (HSCT) has been the primary treatment for RD since its first successful application in 1983. While initially, transplants were performed without preparative conditioning, it was later found that myeloablative conditioning is necessary for successful myeloid function reconstitution. Most RD patients received transplants from either HLA-haploidentical family donors or HLA-matched unrelated donors due to the scarcity of HLA-identical sibling donors. Although HSCT carries significant risks, its implementation is crucial for RD patients due to the high risk of fatal infections. Long-term survivors of HSCT demonstrate the therapy’s potential to permanently restore normal blood cell and immune system functions and cure RD.
References
-
Fujisawa K, Murakami R, Horiguchi T, Noma T. “Adenylate kinase isozyme 2 is essential for growth and development of Drosophila melanogaster.” Comp Biochem Physiol B Biochem Mol Biol. 2009 Jan 18. [Epub ahead of print]
-
Gitlin D, Vawter G, Craig JM. “Thymic alymphoplasia and congenital aleukocytosis.” Pediatrics 1964; 33: 184–192.
-
Haas RJ, Niethammer D, Goldmann SF, et al. “Congenital immunodeficiency and agranulocytosis (reticular dysgenesia).” Acta Paediatr Scand 1977; 66: 279–283.
-
Heltzer ML, Paessler M, Raffini L, et al. “Successful haploidentical bone marrow transplantation in a patient with reticular dysgenesis: three-year follow-up.” J Allergy Clin Immunol 2007; 120: 950–952.
-
Lagresle-Peyrou C, Six EM, Picard C, et al. “Human adenylate kinase 2 deficiency causes a profound hematopoietic defect associated with sensorineural deafness.” Nat Genet 2009; 41: 106–111.
-
Lee HJ, Pyo JO, Oh Y, et al. “AK2 activates a novel apoptotic pathway through formation of a complex with FADD and caspase-10.” Nat Cell Biol 2007; 9: 1303–1310.
-
Levinsky RJ, Tiedeman K. “Successful bone-marrow transplantation for reticular dysgenesis.” Lancet 1983; 1: 671–673.
-
Müller SM, Ege M, Pottharst A, et al. “Transplacentally acquired maternal T lymphocytes in severe combined immunodeficiency: a study of 121 patients.” Blood 2001; 98: 1847–1851.
Share


Tags
Counters